Teich Lab
Teich Lab

Our group has an ongoing collaboration with Dr. Guy McKhann and colleagues to better characterize the effect of Alzheimer’s disease (AD) pathology in normal pressure hydrocephalus (NPH) patients. Cortical shunting of NPH patients involves removal of a brain biopsy at the shunt insertion point at many centers, and early AD pathology occurs with some frequency in these biopsies.
Several groups have used this patient population to study the pathophysiology of AD, the major advantage being that the AD pathology (when it does occur) is generally in the early stages, brain tissue and biofluids can be gathered at the same time point and without post-mortem artifact, and patients can be followed prospectively.
Our recent publication (Huang et al.) involves an analysis of bulk RNA-seq on 106 cortical biopsies that were taken during shunt placement for normal pressure hydrocephalus with varying degrees of comorbid AD pathology. A restricted set of genes correlate with AD pathology in these biopsies, and co-expression network analysis demonstrates an evolution from microglial homeostasis to a disease-associated microglial phenotype in conjunction with increasing AD pathologic burden, along with a subset of additional astrocytic and neuronal genes that accompany these changes. Ongoing work is further evaluating this early response and its relationship with clinical features of AD.
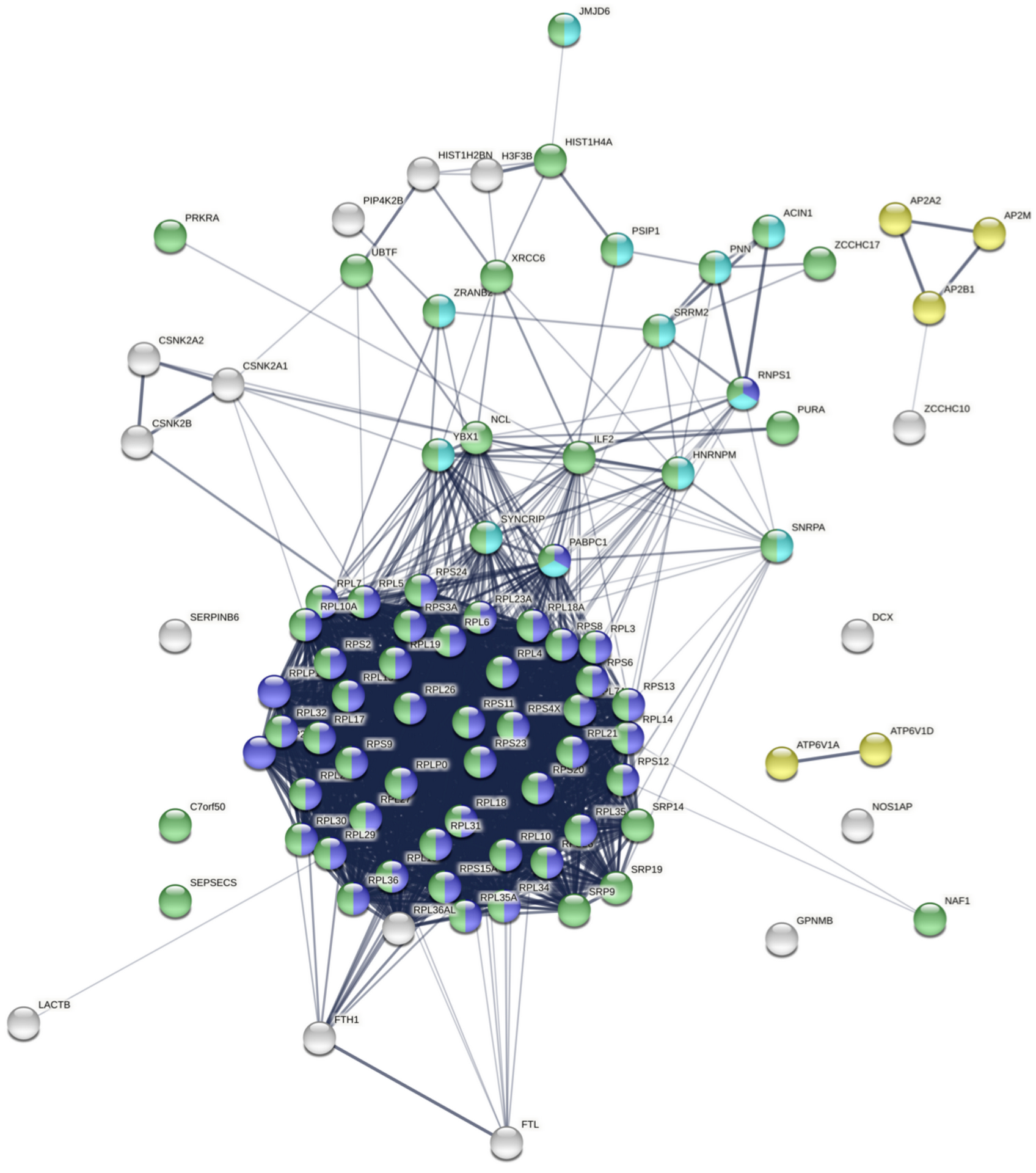
Our group has a long-standing interest in how dysregulation of gene expression in AD contributes to synaptic dysfunction. In Tomljanovic et al., we analyzed neuronal gene expression data from human AD brain tissue to identify master regulators of synaptic gene expression.
We identified ZCCHC17 loss as a driver of synaptic dysfunction through dysregulation of synaptic gene expression. We subsequently validated ZCCHC17 loss in AD brain tissue, and validated our predictions of the transcriptomic effects of ZCCHC17 loss in a rat neuronal culture knock-down model. In Bartosch et al., we show that ZCCHC17 is an important regulator of gene splicing, and that ZCCHC17 loss in a human iPSC neuron model reproduces splicing abnormalities seen in AD brain tissue (including dysregulated synaptic gene splicing). We also show that ZCCHC17 levels in AD patients are predictive of cognitive resilience, and identify tau as a possible mediator of ZCCHC17 dysfunction in AD.
More recent work has demonstrated that ZCCHC17 knock-down phenocopies loss of synaptic proteins and neuronal hyperexcitability seen in AD. In ongoing work, we have also expanded our focus and applied the computational tools used in the above work to a drug screen of over 1000 compounds to identify therapeutic candidates based on modulation of the neuronal transcriptome.